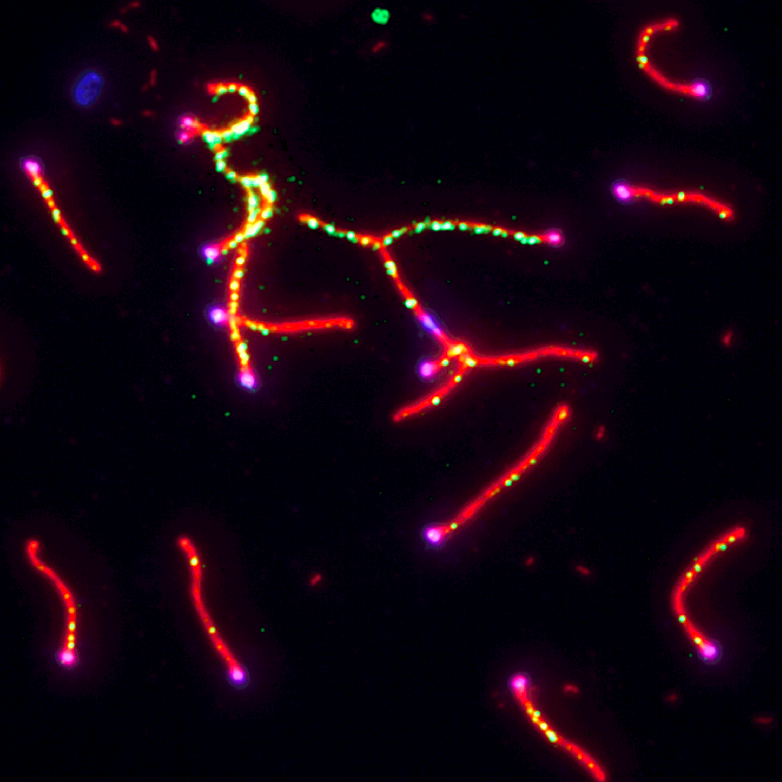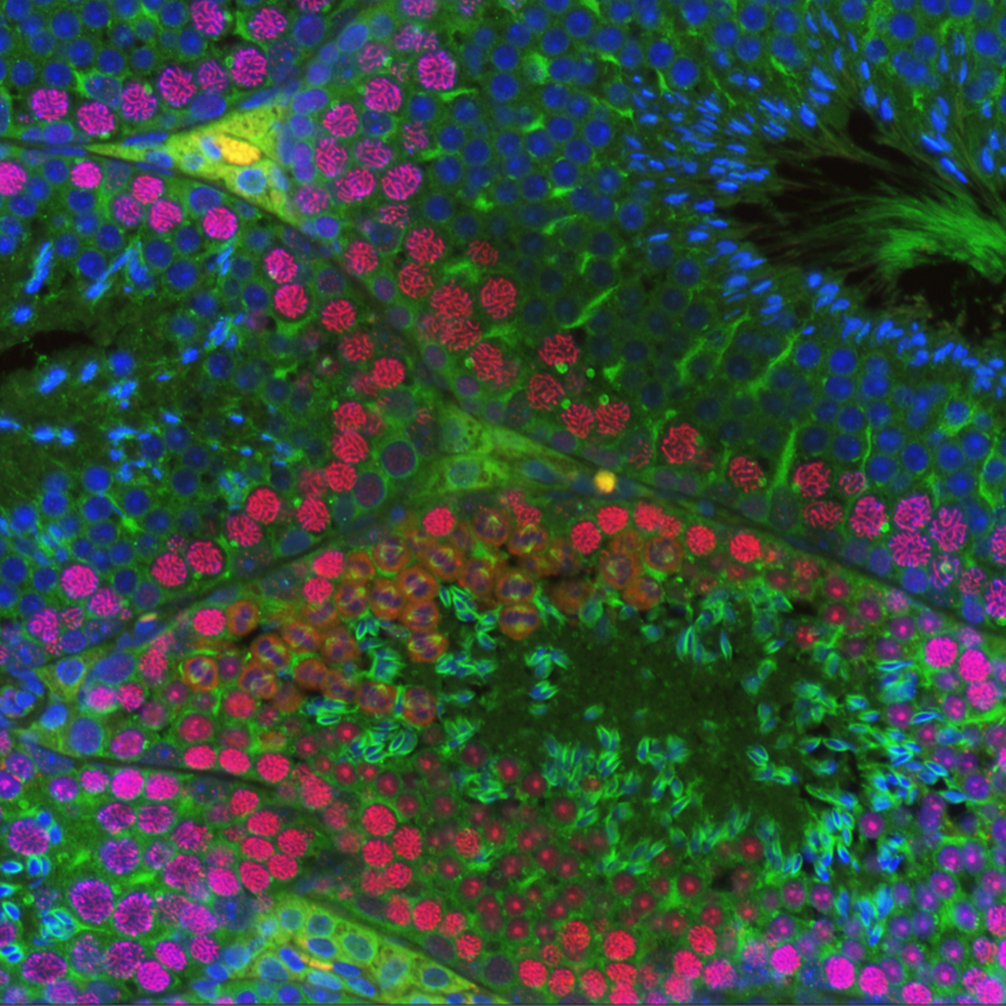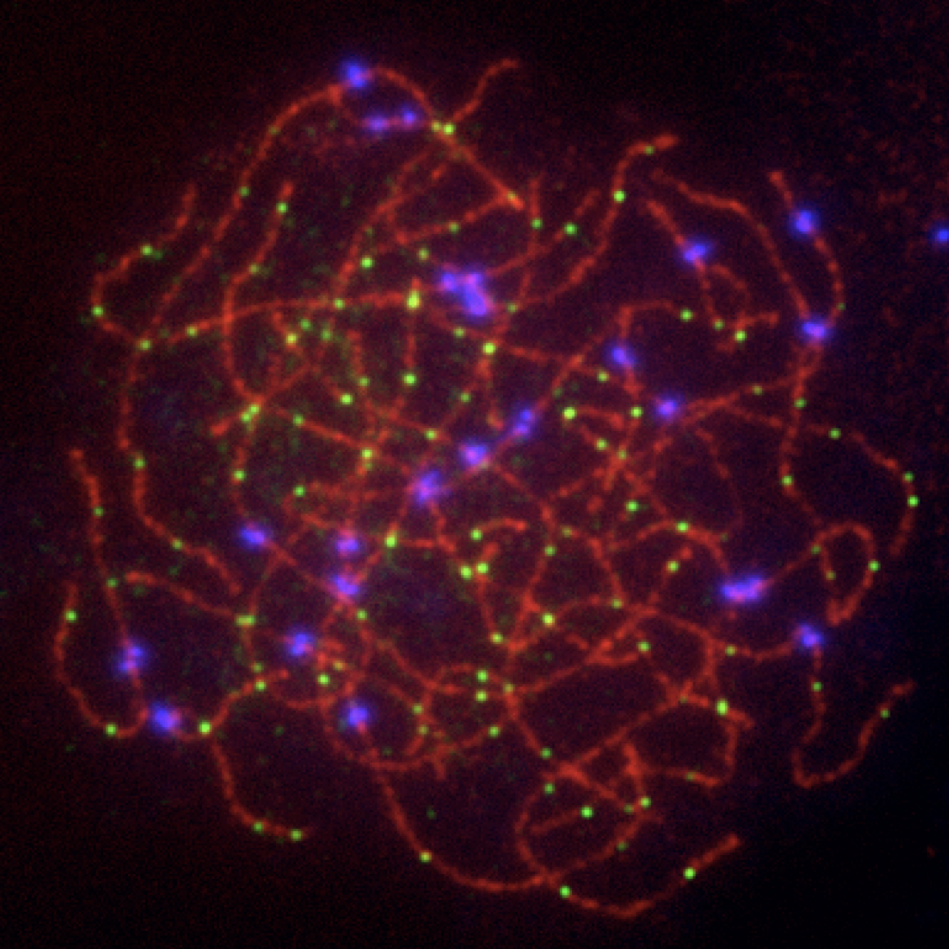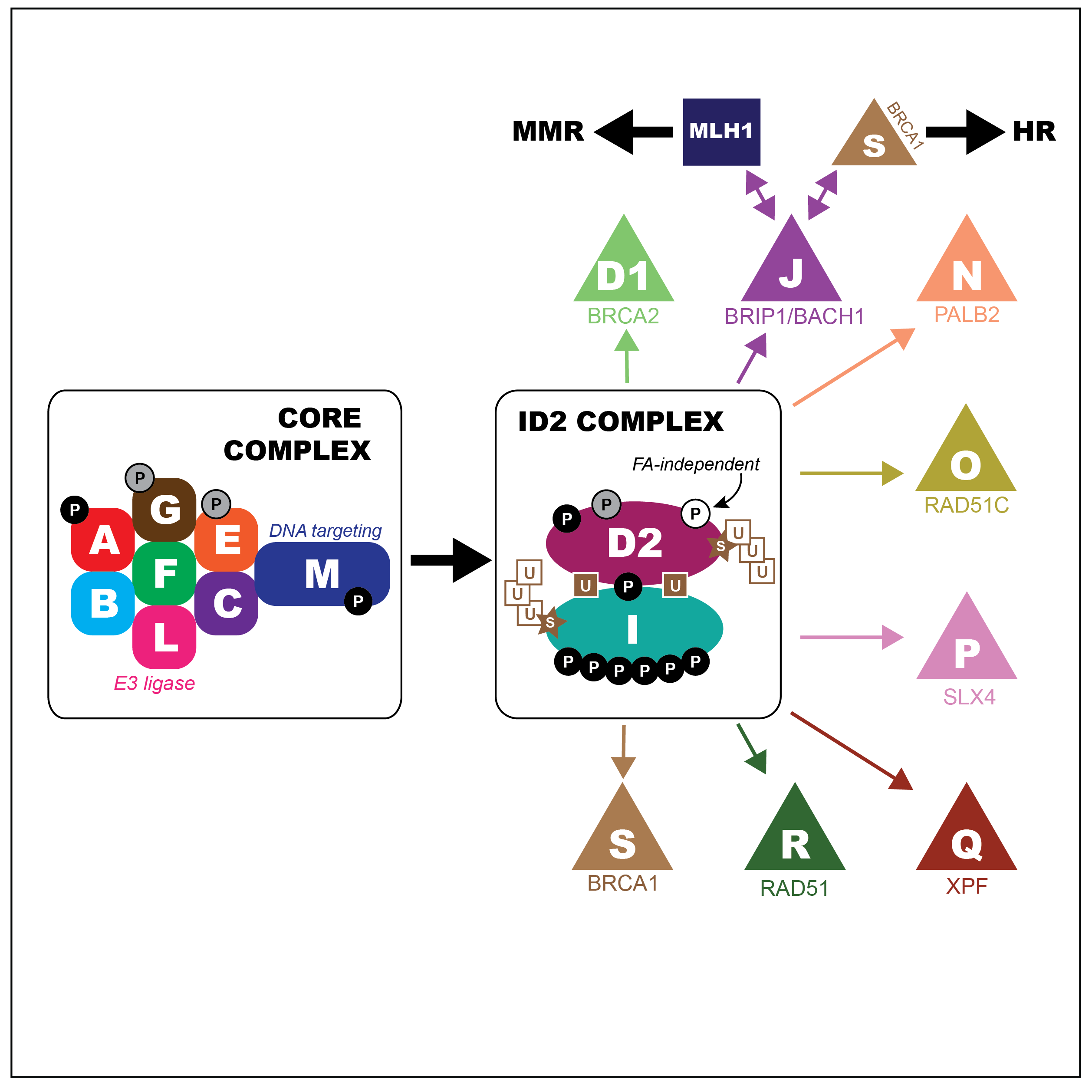
Fanconi anemia (FA) is a rare congenital disorder associated with heightened sensitivity to DNA damaging agents and cancer predisposition. The FA network of proteins act as a regulatory hub capable of bridging several DSB repair pathways including homologous recombination. Of particular interest to our studies are the scaffold protein SLX4 (BTBD12/FANCP) and the helicase FANCJ (BRIP1). Although each protein interacts with differing DSB repair machinery (SLX4 with MUS81, FANCJ with MLH1 and BLM), our previous research indicated the two may be functionally interdependent. FancJ and Slx4 deficient mice lose expression of SLX4 and FANCJ (respectively) in the germline (Holloway et al, 2011; Sun et al, 2016). Knockouts of either result in an increase of MLH1-marked crossovers; however, only loss of FANCJ causes an increase in overall crossover number. These findings suggest that both FANCJ and SLX4 serve as a locus for crosstalk between the class I and II crossover pathways.
Our continued research focuses on detangling the complex interplay between MUS81, BLM, SLX4, and FANCJ. Using coordinated conditional knockouts, we are probing functional interactions of SLX4 and FANCJ with class I and II crossover machinery. We are also working to identify specific partners interacting with MUS81/SLX4/FANCJ and characterize structural domains necessary for their interactions. Finally, in extending these studies to encompass both male and female meiosis, our research will further elucidate sexual dimorphism in DSB repair and crossover formation.
Summary prepared by Dr. Tegan Horan